- CINÉTIQUE CHIMIQUE
- CINÉTIQUE CHIMIQUEÉtant donné un système chimique comprenant des composés chimiques, dans un état physique quelconque et répartis selon une localisation quelconque, on dira que ce système est chimiquement inerte si aucun des composés présents ne se transforme dans sa nature chimique. Dans le cas contraire, on est en présence d’un système réactionnel qui subit une transformation chimique. Si les composés se transforment dans un rapport fixe et défini en un système de produits, on dira que la transformation ne comporte qu’une réaction chimique.Le but premier de la chimie, minérale ou organique, fut longtemps et demeure souvent l’identification, l’isolement des composés chimiques obtenus lors des transformations; l’objectif est alors de connaître tous les composés auxquels peut conduire la chimie, et le mode opératoire et le déroulement même de la transformation ne sont pris en considération qu’en tant que recette de synthèse. L’étude approfondie de la transformation, du «mouvement», de la structure de la matière, tel fut le centre d’intérêt d’un certain nombre de chimistes, qui développèrent cet aspect de la chimie qu’est la cinétique chimique. En un sens, la cinétique chimique regroupe tous les travaux ayant pour finalité de décrire qualitativement ou quantitativement l’évolution des systèmes chimiques, de comprendre le mécanisme selon lequel, au niveau de la molécule, s’opère le réarrangement des liaisons chimiques qui conditionne la réaction, d’expliciter les forces motrices ou antagonistes qui déterminent cette évolution. Notons ici que certains auteurs usent de l’expression «dynamique chimique» dans un sens équivalent à celle de cinétique chimique. Le domaine d’application de la cinétique s’étend à toute évolution de la matière, qu’elle soit très lente ou très rapide, mettant en cause des modifications d’espèces chimiques au sein de celles-ci. Ainsi, prenant comme objet d’étude le système chimique que constitue la matière des espaces intersidéraux, elle explique que les molécules d’hydrogène y sont en équilibre avec des atomes d’hydrogène; ces atomes, qui dans les conditions normales se recombinent instantanément, ont, en effet, dans les conditions spatiales où la concentration des espèces chimiques se trouve réduite à 104 par litre, des durées de vie qui se chiffrent par milliers d’années, c’est-à-dire qu’ils ne réagissent statistiquement qu’après des durées de cet ordre. Inversement, il est nécessaire de pouvoir étudier des réactions ultra-rapides, recombinaisons de radicaux, neutralisations acide-base, car ce sont des étapes élémentaires des transformations chimiques. Comme le soulignait le professeur M. Eigen dans son allocution de prix Nobel 1967, ces réactions étaient considérées, encore en 1949, comme évoluant à une «vitesse immesurable». Elles ont été mesurées et étudiées, bien que la transformation des réactifs n’y nécessite que des durées de l’ordre de 10-10 seconde.Les objectifs de l’analyse cinétique, quoique scientifiques, débouchent sur l’action. Le but de cette science est, en effet, d’observer les transformations pour parvenir d’abord à les décrire, ensuite à énoncer leurs lois, enfin à obtenir les moyens qui permettent de les contrôler. Or la maîtrise de la nature suppose l’aptitude à freiner, à accélérer ou à orienter sa transformation; accélérer une réaction, c’est ce que l’on veut faire dans un réacteur chimique; freiner une transformation, c’est pouvoir stabiliser un matériau ou stocker un produit. Ainsi, de l’éthylène pourra d’abord être stocké dans un réservoir parce que l’on a déterminé des inhibiteurs qui empêchent sa dégradation, puis polymérisé en ajoutant un catalyseur qui provoque sa transformation sélective en polyéthylène, celui-ci étant à nouveau stabilisé par des additifs qui empêcheront sa dégradation par l’oxygène, l’ozone, la lumière, etc. En outre, parce que quantitative, la cinétique permet le travail de l’ingénieur dans des domaines variés. Sachant par exemple calculer la fréquence et l’exothermicité des recombinaisons de radicaux ou d’ions qui, lors de la rentrée d’un satellite dans l’atmosphère, échaufferont sa paroi, on pourra en tenir compte pour le calcul de la protection de ses parois.L’apparition de la cinétique chimique fut postérieure à l’essor de la chimie du XVIIIe siècle et les premières observations cinétiques peuvent être attribuées à G. Kirchhoff (sur la vitesse d’hydrolyse de l’amidon, 1812) et L. J. Thenard (sur la décomposition de l’eau oxygénée, 1818); la première loi cinétique n’apparaît cependant qu’en 1850 quand Wilhelmy montre que la vitesse d’inversion du glucose est proportionnelle à sa concentration: c’est la loi d’«ordre un»; la loi d’«ordre deux» apparaît avec les travaux d’Harcourt et Esson sur l’action de l’acide iodhydrique sur l’eau oxygénée (1858). Vient ensuite la mise en évidence de principes plus généraux: C. M. Guldberg et P. Waage (1867) montrent qu’un équilibre entre deux formes chimiques est un phénomène dynamique qui traduit l’égalité des vitesses directe et inverse de conversion, et ces travaux sont complétés par ceux de J. H. Vant’ Hoff sur la variation des équilibres en fonction de la température (1885). En 1889, S. Arrhenius énonce la loi de variation des vitesses en fonction de la température, tandis que M. Berthelot étudie la thermochimie des réactions. Dès lors se développent des travaux montrant la complexité réelle des systèmes chimiques. S. Arrhenius propose la théorie des ions (1900); M. E. A. Bodenstein étudie systématiquement la synthèse de l’acide bromhydrique (1907), et ses résultats cinétiques sont interprétés par la présence de radicaux; W. Ostwald dégage la notion de catalyseur (1909). Puis viennent les travaux de Michaelis sur la catalyse enzymatique (1913), de J. N. Brønsted sur la catalyse par les acides (1923). L’essor de la cinétique chimique s’est poursuivi ensuite par une mise en évidence de plus en plus précise de la complexité des transformations réelles pour un contrôle de plus en plus grand de leur vitesse. De ce fait, elle est devenue une discipline de base commune à tous les chimistes. Bien qu’elle soit souvent présentée par son intervention relative à chaque branche de la chimie dans les traités de chimie physique organique, chimie physique du solide, biologie moléculaire ou chimie industrielle, nous la présenterons ici en tant que discipline homogène, indépendante du système chimique auquel on l’applique, afin de souligner l’unité de la chimie et l’unité des lois qui régissent les transformations chimiques.1. Activation de la transformation chimiqueLa compréhension du mouvement d’un système mécanique suppose que l’on connaisse la notion de l’énergie potentielle de gravité qu’il possède du fait même qu’il se trouve soumis à l’influence d’un champ de gravité; un système mécanique statique est souvent métastable; il devrait évoluer par réduction de son énergie potentielle, c’est-à-dire avec libération possible d’énergie, mais en est empêché par des forces appelées réactions.En présence d’un système chimique, la première question est de savoir s’il est susceptible d’évolution et sous quelle influence. L’expérience a permis de dégager les lois de la «thermodynamique chimique», dont nous ne retiendrons ici que la conclusion. Il existe une énergie potentielle chimique, que l’on peut calculer pour tout système, et l’évolution chimique spontanée de celui-ci n’est possible que si elle correspond à une diminution de cette énergie, c’est-à-dire à une libération d’énergie. Un système inerte est très rarement stable, mais le plus souvent métastable et peut, si les conditions nécessaires pour provoquer sa transformation sont réunies, évoluer vers l’un des nombreux systèmes chimiques, d’énergie inférieure à la sienne, que l’on peut envisager de former à partir des mêmes atomes.Le premier but de l’étude cinétique d’un système chimique est d’identifier les moyens de lui faire quitter son état métastable; ce sont les moyens d’activation .L’activation thermiqueLe moyen le plus direct d’activer un système inerte consiste à le porter à une température plus élevée. On exalte alors les mouvements intramoléculaires et intermoléculaires provoquant ainsi, à partir d’un seuil de température, la transformation chimique du système. C’est le cas par exemple pour le mélange méthane-air, dont la combustion ne commence que lorsque sous l’action d’une allumette on en a porté la température, en un point, à une valeur minimale de l’ordre de 300 0C. La combustion dégage ensuite beaucoup d’énergie; le système était donc bien métastable. En outre, il n’y a pas de relation simple entre l’énergie dégagée et la température à laquelle on peut amorcer la réaction; ainsi, la neutralisation d’un acide faible par une base (acide acétique plus soude, par exemple), quoique dégageant beaucoup moins d’énergie, aura lieu à température ambiante.L’inconvénient majeur de l’activation thermique est qu’un système réactionnel, un tant soit peu complexe, conduira, si on le porte à haute température, à toute espèce de réactions, car presque tous les liens interatomiques seront activés de façon non différenciée. On dit que l’activation thermique n’est pas sélective. En revanche, le fait de porter un système à haute température peut rendre métastable un système stable, car le potentiel chimique varie avec la température, mais de façon différente pour les différents composés chimiques. De ce fait, certaines transformations, celles de l’éthane en éthylène ou acétylène par exemple, ne peuvent être effectuées qu’à haute température.L’activation chimiqueUne autre voie pour activer un système chimique consiste à lui ajouter un ou plusieurs constituants chimiques supplémentaires, qui suffiront à transformer un système inerte en système évolutif. On parlera alors d’activation chimique. S’il suffit d’introduire ces composés en très faible quantité, et si on les retrouve non modifiés après la transformation du système, on dira que ce sont des catalyseurs et que l’on a réalisé une activation catalytique.La conservation du catalyseur ne signifie pas son inertie; en fait, le catalyseur intervient pour adsorber ou complexer ou même pour réagir avec une ou plusieurs des espèces chimiques présentes. L’espèce chimique ainsi modifiée réagit avec d’autres constituants du système, mais le catalyseur se trouve ramené à son état initial par un acte chimique, inverse de l’acte d’activation. Le catalyseur est donc modifié et régénéré par un mécanisme cyclique. Suivant leur nature et leurs caractéristiques, les catalyseurs activeront préférentiellement telle ou telle espèce chimique et orienteront sélectivement la transformation du système vers tel ou tel des états thermodynamiquement possibles. Suivant la nature physique, chimique ou l’origine des catalyseurs, on distingue les catalyseurs hétérogènes et les catalyseurs homogènes, catalyseurs acide-base ou catalyseurs par complexes de coordination, et enfin les catalyseurs enzymatiques.Dans d’autres cas, l’activation du système n’est obtenue qu’en engageant le constituant supplémentaire en grande quantité, par exemple comme diluant d’un système gazeux ou comme solvant d’un système soluble. Si ce composé n’est pas transformé lors de la réaction qu’il provoque, on parlera d’activation par solvant. Celle-ci peut être très importante, ainsi la réaction d’une amine sur un iodure, par exemple:
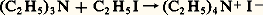 se déroule très lentement dans le cyclohexane, mais mille fois plus vite dans le benzène, cinquante mille fois plus vite dans le nitrobenzène. L’action du solvant sera plus efficace sur certaines espèces que sur d’autres, elle peut donc être sélective.Un troisième cas d’activation chimique correspond à l’introduction dans le système d’un composé chimique en très faible quantité, qui provoque la transformation du système, mais est, contrairement au catalyseur, consommé lors de la réaction. C’est ainsi que l’addition d’un kilogramme d’ester peroxydicarbonique à de l’éthylène, condensé à 200 0C sous une pression de 1 000 kg/cm2, permet d’en polymériser deux tonnes en polyéthylène. On parle alors d’initiateur chimique et d’initiation chimique .Les activations physiquesLe fait de soumettre un système chimique à l’action d’un rayonnement corpusculaire, ou plus généralement de l’onde associée, permet dans certains cas de l’activer. C’est ainsi que l’on peut rompre des macromolécules par l’action d’ultrasons, mais le plus souvent il faut choisir des ondes de fréquence plus élevée, c’est-à-dire susceptibles d’apporter plus d’énergie. L’activation photochimique, qui correspond à l’action des ondes lumineuses, surtout ultraviolettes, est la plus utilisée car la mieux adaptée. Cela n’a rien de surprenant puisque l’émission même des ondes, dans ce domaine de longueur d’onde, correspond à l’émission d’énergie provoquée par le passage d’un électron d’une orbitale à une autre orbitale d’énergie plus basse. L’activation consiste dans le phénomène inverse, l’énergie du quanta lumineux permettant à un électron de passer d’une orbitale à une orbitale plus élevée; l’activation ainsi obtenue peut être très sélective. D’autre part, l’apport d’énergie peut servir soit à initier une réaction (c’est le cas des plaques photographiques où très peu d’énergie provoque une transformation notable), soit réellement à apporter l’énergie nécessaire à l’évolution d’un système stable: c’est le cas de la synthèse chlorophyllienne où la lumière permet la réaction du système stable constitué par le gaz carbonique et l’eau (CO2 + H2O).Les ondes électromagnétiques de plus haute énergie, rayonnement 塚 d’une source radio-isotopique, rayons X, rayonnements électroniques ou corpusculaires, peuvent être utilisées. L’énergie qu’elles apportent est souvent beaucoup trop grande et active indifféremment toutes les espèces chimiques. Son action est donc peu sélective.L’activation électrochimiqueAlors que l’utilisation d’électrons de haute énergie peut être assimilée au cas précédent par ses défauts (effluves électriques), l’activation par électron peut être réalisée de façon contrôlée et sélective par l’utilisation de l’électrochimie, l’électron étant introduit dans le système à la cathode où il réduit un composé A (A + e 藺-), tandis qu’il est repris à l’autre électrode, l’anode, où un composé B du système est oxydé (B隣+ + e ).Les électrodes s’apparentent à des catalyseurs hétérogènes permettant l’utilisation, elle aussi catalytique, de l’électron. Mais une caractéristique importante réside dans le fait que l’énergie chimique de la réaction (A + B藺- + B+) est aussi convertible en énergie électrique. On peut soit apporter de l’énergie au système, et provoquer alors la réaction de systèmes stables (électrolyse de l’alumine, du chlorure de sodium), ou inversement ressortir sous forme d’énergie électrique l’énergie d’une réaction chimique (piles à combustibles).Ces moyens d’activation ne sont pas équivalents: ils se distinguent par leurs conditions de mise en œuvre, leur efficacité et leur sélectivité. Ils permettent de rendre réactionnel presque tout système chimique, mais dans la pratique on devra choisir celui qui conduit à la meilleure économie. Cela supposera une connaissance fine de leur action pour l’optimaliser et la comparer. S’ils se révèlent trop coûteux, on aura parfois intérêt à faire subir au système chimique des modifications chimiques préalables par des réactifs supplémentaires pour le transformer en un système se prêtant mieux à une attaque sélective.2. Localisation de la transformation chimiqueUne donnée importante sur la transformation chimique est sa localisation. Il y a d’ailleurs lieu tout d’abord de réexaminer la notion de système chimique: celui-ci est défini comme un certain volume de matière que l’on doit pouvoir, pour l’étudier en tant que tel, limiter par une enveloppe dont de préférence l’interaction chimique avec le système est nulle. Cela est possible assez souvent pour des systèmes évoluant à température faible grâce à des matériaux inertes, verre, «inox», voire titane. Quand cela n’est pas possible, et c’est le cas général pour les hautes températures parce que les parois «réfractaires» sont souvent chimiquement impliquées dans le système, on doit choisir une frontière fictive du système, frontière au travers de laquelle on devra mesurer les transferts de matière relatifs à tous les constituants chimiques du système; c’est le cas par exemple des fronts de flamme ou des réactions dans les plasmas ionisés, systèmes chimiques où l’on parvient à réaliser des températures de 3 000 0C ou largement supérieures.Il est également primordial pour la conception des mesures, et pour l’interprétation des résultats, de savoir si la réaction se produit ou non de manière uniforme dans une phase homogène. Une telle réaction est dite homogène au sens strict. Ce cas est très favorable, puisque la mesure, en un point, informe sur l’ensemble, et on essaiera de le réaliser dans les études en laboratoire bien qu’il soit en fait assez exceptionnel dans la pratique. En effet, même dans des systèmes ne comportant qu’une seule phase au sens physico-chimique du terme, les écarts de composition seront souvent tels que, du point de vue cinétique, il faudra l’étudier comme un système non uniforme, c’est-à-dire déterminer l’état et les concentrations des espèces chimiques en tout point du système. Tel est le cas pour l’évolution d’une masse gazeuse ou liquide si la réaction est initiée, ou inhibée, par la paroi du récipient, phénomène fréquent qui se traduit par la sensibilité de ces systèmes à la nature des parois; par exemple, la décomposition d’eau oxygénée est très sensible au matériau de stockage utilisé.Les systèmes hétérogènes sont les plus fréquents, la quantité des cas possibles étant très grande par le nombre et la nature des phases présentes, le degré d’uniformité de chacune d’elles, et surtout par la variété des facteurs cinétiques qui exercent leur influence en tout point du système. Un cas souvent rencontré est celui d’un système biphasique liquide-gaz; la réaction sera homogène dans chacune des phases, pour autant que l’équilibre de composition entre les phases puisse être assuré en permanence. Lorsque les phénomènes de transfert de matière à l’interface deviennent trop lents, il se produit des gradients de concentration à son voisinage et le phénomène est plus complexe à suivre. L’étude des réactions hétérogènes devient plus délicate encore si, outre les phases fluides, il intervient une ou plusieurs phases solides. Le comportement de ces phases solides sera déterminé dans une large mesure par leur structure et leur texture, mais aussi par les caractéristiques de l’interface qui peut, lui, être affecté par des imperfections du réseau du solide, et qu’il faudrait pouvoir connaître au niveau même de la position des atomes; or, les méthodes physiques sont disponibles pour connaître les compositions ou les caractéristiques structurales des phases, mais sont souvent insuffisantes pour les études de surface. Dans le cas, très fréquent en chimie organique industrielle, où le système chimique est constitué par une phase fluide dont la transformation est catalysée par un solide, la difficulté est de nature semblable, mais simplifiée toutefois par le fait que, le catalyseur évoluant peu, le front de réaction reste constant en forme, en étendue et en quantité. Il y aura de toute façon avantage dans tous ces cas à uniformiser les phases fluides par une agitation intense. On pourra alors au moins considérer que la composition au sein de la phase fluide est également celle qui existe au voisinage de l’interface catalytique.Un cas encore plus difficile de localisation apparaît en biochimie au fur et à mesure que l’on veut étudier la cinétique des transformations biochimiques au niveau moléculaire et in vivo. Il apparaît alors que ce qui, il y a cinquante ans, sous observation au microscope, pouvait encore passer pour une phase uniforme est en fait un système chimique où la position de toutes les macromolécules, acides nucléiques, enzymes, polysaccharides, est fixe, les petites molécules, eau, cations inorganiques, acides aminés, étant plus ou moins libres. On doit donc avoir une description «topochimique» de l’ensemble de la cellule pour comprendre les transformations qui s’y déroulent. Cette non-uniformité s’étend même au niveau des maillons des polymères. Une molécule d’acide ribonucléique, élément génétique de la cellule, doit avoir un enchaînement absolument défini des maillons élémentaires pour conserver l’information relative aux caractéristiques de la cellule; la compréhension des mécanismes chimiques liés à la génétique suppose là aussi la connaissance de l’emplacement relatif des divers gènes, de la carte génétique. C’est l’hétérogénéité poussée au niveau de la localisation même des molécules qui permet l’efficacité des systèmes biochimiques, mais rend l’étude cinétique des transformations qui s’y déroulent très difficile.3. Formes réelles des espèces chimiquesConstituer un système chimique, par mélange de plusieurs composés chimiques, est considéré, si les espèces chimiques demeurent récupérables par fractionnement du système, comme une simple opération physique; en fait, il se produit souvent une organisation entre les molécules des espèces chimiques engagées, véritable transformation chimique conduisant à de nouvelles espèces; c’est parce que cette transformation est aisément réversible qu’elle est annulée lors du fractionnement du système et que l’on a eu tendance à la négliger alors qu’elle est d’une grande importance pour comprendre les transformations chimiques que subirait le système si on l’activait. L’étude in situ des systèmes chimiques, sans isolement des constituants chimiques, rendue possible par le développement des méthodes spectroscopiques (infrarouge, ultraviolet, résonance magnétique nucléaire, résonance paramagnétique électronique) a permis l’étude de ces formes.Ainsi un composé salin AB, chlorure de sodium par exemple, est sous forme cristalline une organisation structurée d’ions Cl- et Na+. Mis en solution dans l’eau, ces ions seront rendus indépendants, mais tous seront solvatés par des molécules d’eau (Cl-, n H2O) et (Na+, n H2O). Dans un solvant organique, de constante diélectrique plus faible que l’eau, l’alcool par exemple, les ions se neutraliseront deux à deux et seront toujours couplés à l’état de paire d’ions (Cl-, Na+). De même l’acide acétique, CH3C2H, existe le plus souvent sous forme auto-associée, à l’état de dimères ouverts ou fermés correspondant aux structures décrites dans la figure 1. Ces formes, en équilibre entre elles, représentent la nature réelle de l’espèce chimique dans un système qui peut être inerte ou réactif. Dans ce dernier cas, la commutation des liaisons interatomiques que suppose la transformation irréversible des espèces s’opère par des modes variés, mais nécessite le passage des réactifs par des formes intermédiaires , parfois extrêmement fugaces, et de ce fait présentes en très faible concentration dans le système. La nature de ces formes actives , qui peuvent être, dans des cas de plus en plus nombreux, observées in situ dans le système en évolution, confère à la transformation certains de ces caractères marquants. On distingue quatre types principaux de formes actives.Les molécules activéesL’état normal d’une molécule correspond à la structure et à la configuration électronique conduisant à l’énergie la plus faible. Si un électron, qui dans l’état stable occupe une certaine orbitale, est promu sur une orbitale de plus haute énergie, la molécule sera activée, c’est-à-dire moins stable ou plus réactive. Ainsi l’oxygène peut exister sous plusieurs formes activées; alors que la forme stable a une configuration électronique où les deux électrons de plus haute énergie ont des spins parallèles (état triplet), beaucoup de réactions supposent que l’oxygène passe par une forme activée où ces deux électrons ont des spins antiparallèles (état singulet). Ces états activés se décèlent surtout par spectroscopie ultraviolette ou par mesure des caractéristiques magnétiques.Les radicauxCertaines formes actives résultent de la scission d’une molécule, chaque fragment conservant un électron du couple d’électrons qui assurait la liaison dans la molécule; c’est une scission homolytique donnant deux radicaux, comportant chacun un électron non apparié. De tels radicaux sont extrêmement réactionnels et permettent en particulier des mécanismes en chaîne. La scission qui les crée est provoquée thermiquement, photochimiquement ou par un initiateur chimique. Ils ont été détectés par les conséquences logiques de la cinétique chimique; aujourd’hui, la résonance paramagnétique électronique permet de les identifier in situ (fig. 2). Notons enfin que le rôle des neutrons dans les phénomènes de fission s’apparente, mais au niveau atomique, au rôle des radicaux dans les réactions en chaîne.Les ionsLa scission d’une molécule peut également se faire de façon hétérolytique, un fragment conservant les deux électrons du lieu chimique; il y a alors formation de deux ions (cation A+, anion B-). Leur formation peut résulter aussi du transfert d’un électron de l’espèce A à l’espèce B (oxydo-réduction). Ces phénomènes sont très fréquents à l’état solide et à l’état liquide, surtout dans les milieux de forte constante diélectrique. En phase gazeuse, les ions n’apparaissent qu’à haute température, mais jouent alors un rôle aussi important que les radicaux. L’identification des ions repose sur des méthodes spécifiques, conductimétrie en phase liquide, spectrographie de masse en phase gazeuse, qui s’ajoutent aux méthodes spectroscopiques usuelles.Les complexesLa forme la plus fréquente d’organisation des molécules résulte de la formation de complexes par association d’espèces chimiques. Ainsi, dans le cas d’un solide plongé dans une phase fluide, la surface du solide sera le plus souvent occupée par des molécules des espèces de la phase fluide, qui sont complexées, adsorbées sur le solide. De même un cation A+ sera le plus souvent complexé, solvaté, par des molécules polaires ayant tendance à donner des électrons (électrodonneurs), et inversement pour un anion. Les complexes se formeront également très souvent entre molécules neutres, en particulier par pont hydrogène. C’est dans le domaine de la biochimie que la formation de complexes fut le plus tôt et le plus nettement revendiquée (Michaelis, 1913), et c’est là qu’elle joue le rôle le plus fin, puisque beaucoup des phénomènes spécifiques s’expliquent par le fait qu’une macromolécule biochimique ne peut complexer qu’une espèce chimique bien définie ayant exactement les caractéristiques complémentaires du site de complexation (fig. 1 b).Les formes actives sont d’autant plus difficiles à observer qu’elles sont plus réactives. C’est la raison pour laquelle leur existence a souvent été établie par le raisonnement à partir des données de la cinétique. Cela reste encore souvent vrai, car les méthodes physiques maintenant disponibles pour l’observation in situ révèlent en premier les formes ions, complexes, en forte concentration, c’est-à-dire des formes stables résultant d’équilibres réversibles, et c’est parmi ces espèces qu’il faudra déceler les formes, peu stables, donc en faible concentration, que sont les formes actives de la transformation. Cette difficulté a souvent conduit à de fausses interprétations.4. Cinétique formelleLa description de l’évolution du phénomène chimique doit être poussée jusqu’à pouvoir être quantitative. Après que la nature des constituants a été identifiée, les mesures quantitatives qui serviront à décrire le système sont des mesures de nombre de molécules dans le système, Ni pour un constituant i.Les données fondamentales pour la description en cinétique chimique des phénomènes sont des vitesses d’apparition ou de disparition des constituants par unité de temps et de volume ( 律 désigne le volume du système considéré):
se déroule très lentement dans le cyclohexane, mais mille fois plus vite dans le benzène, cinquante mille fois plus vite dans le nitrobenzène. L’action du solvant sera plus efficace sur certaines espèces que sur d’autres, elle peut donc être sélective.Un troisième cas d’activation chimique correspond à l’introduction dans le système d’un composé chimique en très faible quantité, qui provoque la transformation du système, mais est, contrairement au catalyseur, consommé lors de la réaction. C’est ainsi que l’addition d’un kilogramme d’ester peroxydicarbonique à de l’éthylène, condensé à 200 0C sous une pression de 1 000 kg/cm2, permet d’en polymériser deux tonnes en polyéthylène. On parle alors d’initiateur chimique et d’initiation chimique .Les activations physiquesLe fait de soumettre un système chimique à l’action d’un rayonnement corpusculaire, ou plus généralement de l’onde associée, permet dans certains cas de l’activer. C’est ainsi que l’on peut rompre des macromolécules par l’action d’ultrasons, mais le plus souvent il faut choisir des ondes de fréquence plus élevée, c’est-à-dire susceptibles d’apporter plus d’énergie. L’activation photochimique, qui correspond à l’action des ondes lumineuses, surtout ultraviolettes, est la plus utilisée car la mieux adaptée. Cela n’a rien de surprenant puisque l’émission même des ondes, dans ce domaine de longueur d’onde, correspond à l’émission d’énergie provoquée par le passage d’un électron d’une orbitale à une autre orbitale d’énergie plus basse. L’activation consiste dans le phénomène inverse, l’énergie du quanta lumineux permettant à un électron de passer d’une orbitale à une orbitale plus élevée; l’activation ainsi obtenue peut être très sélective. D’autre part, l’apport d’énergie peut servir soit à initier une réaction (c’est le cas des plaques photographiques où très peu d’énergie provoque une transformation notable), soit réellement à apporter l’énergie nécessaire à l’évolution d’un système stable: c’est le cas de la synthèse chlorophyllienne où la lumière permet la réaction du système stable constitué par le gaz carbonique et l’eau (CO2 + H2O).Les ondes électromagnétiques de plus haute énergie, rayonnement 塚 d’une source radio-isotopique, rayons X, rayonnements électroniques ou corpusculaires, peuvent être utilisées. L’énergie qu’elles apportent est souvent beaucoup trop grande et active indifféremment toutes les espèces chimiques. Son action est donc peu sélective.L’activation électrochimiqueAlors que l’utilisation d’électrons de haute énergie peut être assimilée au cas précédent par ses défauts (effluves électriques), l’activation par électron peut être réalisée de façon contrôlée et sélective par l’utilisation de l’électrochimie, l’électron étant introduit dans le système à la cathode où il réduit un composé A (A + e 藺-), tandis qu’il est repris à l’autre électrode, l’anode, où un composé B du système est oxydé (B隣+ + e ).Les électrodes s’apparentent à des catalyseurs hétérogènes permettant l’utilisation, elle aussi catalytique, de l’électron. Mais une caractéristique importante réside dans le fait que l’énergie chimique de la réaction (A + B藺- + B+) est aussi convertible en énergie électrique. On peut soit apporter de l’énergie au système, et provoquer alors la réaction de systèmes stables (électrolyse de l’alumine, du chlorure de sodium), ou inversement ressortir sous forme d’énergie électrique l’énergie d’une réaction chimique (piles à combustibles).Ces moyens d’activation ne sont pas équivalents: ils se distinguent par leurs conditions de mise en œuvre, leur efficacité et leur sélectivité. Ils permettent de rendre réactionnel presque tout système chimique, mais dans la pratique on devra choisir celui qui conduit à la meilleure économie. Cela supposera une connaissance fine de leur action pour l’optimaliser et la comparer. S’ils se révèlent trop coûteux, on aura parfois intérêt à faire subir au système chimique des modifications chimiques préalables par des réactifs supplémentaires pour le transformer en un système se prêtant mieux à une attaque sélective.2. Localisation de la transformation chimiqueUne donnée importante sur la transformation chimique est sa localisation. Il y a d’ailleurs lieu tout d’abord de réexaminer la notion de système chimique: celui-ci est défini comme un certain volume de matière que l’on doit pouvoir, pour l’étudier en tant que tel, limiter par une enveloppe dont de préférence l’interaction chimique avec le système est nulle. Cela est possible assez souvent pour des systèmes évoluant à température faible grâce à des matériaux inertes, verre, «inox», voire titane. Quand cela n’est pas possible, et c’est le cas général pour les hautes températures parce que les parois «réfractaires» sont souvent chimiquement impliquées dans le système, on doit choisir une frontière fictive du système, frontière au travers de laquelle on devra mesurer les transferts de matière relatifs à tous les constituants chimiques du système; c’est le cas par exemple des fronts de flamme ou des réactions dans les plasmas ionisés, systèmes chimiques où l’on parvient à réaliser des températures de 3 000 0C ou largement supérieures.Il est également primordial pour la conception des mesures, et pour l’interprétation des résultats, de savoir si la réaction se produit ou non de manière uniforme dans une phase homogène. Une telle réaction est dite homogène au sens strict. Ce cas est très favorable, puisque la mesure, en un point, informe sur l’ensemble, et on essaiera de le réaliser dans les études en laboratoire bien qu’il soit en fait assez exceptionnel dans la pratique. En effet, même dans des systèmes ne comportant qu’une seule phase au sens physico-chimique du terme, les écarts de composition seront souvent tels que, du point de vue cinétique, il faudra l’étudier comme un système non uniforme, c’est-à-dire déterminer l’état et les concentrations des espèces chimiques en tout point du système. Tel est le cas pour l’évolution d’une masse gazeuse ou liquide si la réaction est initiée, ou inhibée, par la paroi du récipient, phénomène fréquent qui se traduit par la sensibilité de ces systèmes à la nature des parois; par exemple, la décomposition d’eau oxygénée est très sensible au matériau de stockage utilisé.Les systèmes hétérogènes sont les plus fréquents, la quantité des cas possibles étant très grande par le nombre et la nature des phases présentes, le degré d’uniformité de chacune d’elles, et surtout par la variété des facteurs cinétiques qui exercent leur influence en tout point du système. Un cas souvent rencontré est celui d’un système biphasique liquide-gaz; la réaction sera homogène dans chacune des phases, pour autant que l’équilibre de composition entre les phases puisse être assuré en permanence. Lorsque les phénomènes de transfert de matière à l’interface deviennent trop lents, il se produit des gradients de concentration à son voisinage et le phénomène est plus complexe à suivre. L’étude des réactions hétérogènes devient plus délicate encore si, outre les phases fluides, il intervient une ou plusieurs phases solides. Le comportement de ces phases solides sera déterminé dans une large mesure par leur structure et leur texture, mais aussi par les caractéristiques de l’interface qui peut, lui, être affecté par des imperfections du réseau du solide, et qu’il faudrait pouvoir connaître au niveau même de la position des atomes; or, les méthodes physiques sont disponibles pour connaître les compositions ou les caractéristiques structurales des phases, mais sont souvent insuffisantes pour les études de surface. Dans le cas, très fréquent en chimie organique industrielle, où le système chimique est constitué par une phase fluide dont la transformation est catalysée par un solide, la difficulté est de nature semblable, mais simplifiée toutefois par le fait que, le catalyseur évoluant peu, le front de réaction reste constant en forme, en étendue et en quantité. Il y aura de toute façon avantage dans tous ces cas à uniformiser les phases fluides par une agitation intense. On pourra alors au moins considérer que la composition au sein de la phase fluide est également celle qui existe au voisinage de l’interface catalytique.Un cas encore plus difficile de localisation apparaît en biochimie au fur et à mesure que l’on veut étudier la cinétique des transformations biochimiques au niveau moléculaire et in vivo. Il apparaît alors que ce qui, il y a cinquante ans, sous observation au microscope, pouvait encore passer pour une phase uniforme est en fait un système chimique où la position de toutes les macromolécules, acides nucléiques, enzymes, polysaccharides, est fixe, les petites molécules, eau, cations inorganiques, acides aminés, étant plus ou moins libres. On doit donc avoir une description «topochimique» de l’ensemble de la cellule pour comprendre les transformations qui s’y déroulent. Cette non-uniformité s’étend même au niveau des maillons des polymères. Une molécule d’acide ribonucléique, élément génétique de la cellule, doit avoir un enchaînement absolument défini des maillons élémentaires pour conserver l’information relative aux caractéristiques de la cellule; la compréhension des mécanismes chimiques liés à la génétique suppose là aussi la connaissance de l’emplacement relatif des divers gènes, de la carte génétique. C’est l’hétérogénéité poussée au niveau de la localisation même des molécules qui permet l’efficacité des systèmes biochimiques, mais rend l’étude cinétique des transformations qui s’y déroulent très difficile.3. Formes réelles des espèces chimiquesConstituer un système chimique, par mélange de plusieurs composés chimiques, est considéré, si les espèces chimiques demeurent récupérables par fractionnement du système, comme une simple opération physique; en fait, il se produit souvent une organisation entre les molécules des espèces chimiques engagées, véritable transformation chimique conduisant à de nouvelles espèces; c’est parce que cette transformation est aisément réversible qu’elle est annulée lors du fractionnement du système et que l’on a eu tendance à la négliger alors qu’elle est d’une grande importance pour comprendre les transformations chimiques que subirait le système si on l’activait. L’étude in situ des systèmes chimiques, sans isolement des constituants chimiques, rendue possible par le développement des méthodes spectroscopiques (infrarouge, ultraviolet, résonance magnétique nucléaire, résonance paramagnétique électronique) a permis l’étude de ces formes.Ainsi un composé salin AB, chlorure de sodium par exemple, est sous forme cristalline une organisation structurée d’ions Cl- et Na+. Mis en solution dans l’eau, ces ions seront rendus indépendants, mais tous seront solvatés par des molécules d’eau (Cl-, n H2O) et (Na+, n H2O). Dans un solvant organique, de constante diélectrique plus faible que l’eau, l’alcool par exemple, les ions se neutraliseront deux à deux et seront toujours couplés à l’état de paire d’ions (Cl-, Na+). De même l’acide acétique, CH3C2H, existe le plus souvent sous forme auto-associée, à l’état de dimères ouverts ou fermés correspondant aux structures décrites dans la figure 1. Ces formes, en équilibre entre elles, représentent la nature réelle de l’espèce chimique dans un système qui peut être inerte ou réactif. Dans ce dernier cas, la commutation des liaisons interatomiques que suppose la transformation irréversible des espèces s’opère par des modes variés, mais nécessite le passage des réactifs par des formes intermédiaires , parfois extrêmement fugaces, et de ce fait présentes en très faible concentration dans le système. La nature de ces formes actives , qui peuvent être, dans des cas de plus en plus nombreux, observées in situ dans le système en évolution, confère à la transformation certains de ces caractères marquants. On distingue quatre types principaux de formes actives.Les molécules activéesL’état normal d’une molécule correspond à la structure et à la configuration électronique conduisant à l’énergie la plus faible. Si un électron, qui dans l’état stable occupe une certaine orbitale, est promu sur une orbitale de plus haute énergie, la molécule sera activée, c’est-à-dire moins stable ou plus réactive. Ainsi l’oxygène peut exister sous plusieurs formes activées; alors que la forme stable a une configuration électronique où les deux électrons de plus haute énergie ont des spins parallèles (état triplet), beaucoup de réactions supposent que l’oxygène passe par une forme activée où ces deux électrons ont des spins antiparallèles (état singulet). Ces états activés se décèlent surtout par spectroscopie ultraviolette ou par mesure des caractéristiques magnétiques.Les radicauxCertaines formes actives résultent de la scission d’une molécule, chaque fragment conservant un électron du couple d’électrons qui assurait la liaison dans la molécule; c’est une scission homolytique donnant deux radicaux, comportant chacun un électron non apparié. De tels radicaux sont extrêmement réactionnels et permettent en particulier des mécanismes en chaîne. La scission qui les crée est provoquée thermiquement, photochimiquement ou par un initiateur chimique. Ils ont été détectés par les conséquences logiques de la cinétique chimique; aujourd’hui, la résonance paramagnétique électronique permet de les identifier in situ (fig. 2). Notons enfin que le rôle des neutrons dans les phénomènes de fission s’apparente, mais au niveau atomique, au rôle des radicaux dans les réactions en chaîne.Les ionsLa scission d’une molécule peut également se faire de façon hétérolytique, un fragment conservant les deux électrons du lieu chimique; il y a alors formation de deux ions (cation A+, anion B-). Leur formation peut résulter aussi du transfert d’un électron de l’espèce A à l’espèce B (oxydo-réduction). Ces phénomènes sont très fréquents à l’état solide et à l’état liquide, surtout dans les milieux de forte constante diélectrique. En phase gazeuse, les ions n’apparaissent qu’à haute température, mais jouent alors un rôle aussi important que les radicaux. L’identification des ions repose sur des méthodes spécifiques, conductimétrie en phase liquide, spectrographie de masse en phase gazeuse, qui s’ajoutent aux méthodes spectroscopiques usuelles.Les complexesLa forme la plus fréquente d’organisation des molécules résulte de la formation de complexes par association d’espèces chimiques. Ainsi, dans le cas d’un solide plongé dans une phase fluide, la surface du solide sera le plus souvent occupée par des molécules des espèces de la phase fluide, qui sont complexées, adsorbées sur le solide. De même un cation A+ sera le plus souvent complexé, solvaté, par des molécules polaires ayant tendance à donner des électrons (électrodonneurs), et inversement pour un anion. Les complexes se formeront également très souvent entre molécules neutres, en particulier par pont hydrogène. C’est dans le domaine de la biochimie que la formation de complexes fut le plus tôt et le plus nettement revendiquée (Michaelis, 1913), et c’est là qu’elle joue le rôle le plus fin, puisque beaucoup des phénomènes spécifiques s’expliquent par le fait qu’une macromolécule biochimique ne peut complexer qu’une espèce chimique bien définie ayant exactement les caractéristiques complémentaires du site de complexation (fig. 1 b).Les formes actives sont d’autant plus difficiles à observer qu’elles sont plus réactives. C’est la raison pour laquelle leur existence a souvent été établie par le raisonnement à partir des données de la cinétique. Cela reste encore souvent vrai, car les méthodes physiques maintenant disponibles pour l’observation in situ révèlent en premier les formes ions, complexes, en forte concentration, c’est-à-dire des formes stables résultant d’équilibres réversibles, et c’est parmi ces espèces qu’il faudra déceler les formes, peu stables, donc en faible concentration, que sont les formes actives de la transformation. Cette difficulté a souvent conduit à de fausses interprétations.4. Cinétique formelleLa description de l’évolution du phénomène chimique doit être poussée jusqu’à pouvoir être quantitative. Après que la nature des constituants a été identifiée, les mesures quantitatives qui serviront à décrire le système sont des mesures de nombre de molécules dans le système, Ni pour un constituant i.Les données fondamentales pour la description en cinétique chimique des phénomènes sont des vitesses d’apparition ou de disparition des constituants par unité de temps et de volume ( 律 désigne le volume du système considéré):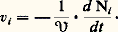 Dans le cas d’un système homogène au sens strict, vi sera identique en tout point et ne dépendra donc que du système et des paramètres physiques globaux, température et pression. Dans les autres cas, il faudra être en mesure de connaître vi en tout point du système soit vi (x , y , z ), la vitesse vi moyenne n’étant que l’intégrale étendue au volume du système des vitesses locales:
Dans le cas d’un système homogène au sens strict, vi sera identique en tout point et ne dépendra donc que du système et des paramètres physiques globaux, température et pression. Dans les autres cas, il faudra être en mesure de connaître vi en tout point du système soit vi (x , y , z ), la vitesse vi moyenne n’étant que l’intégrale étendue au volume du système des vitesses locales: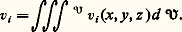 Le cas d’un système isochore sera également plus facile à étudier que celui d’un système évoluant à volume variable.La réaction isoléeL’étude sera surtout beaucoup plus facile si l’on est en présence d’une réaction chimique isolée, c’est-à-dire conduisant à un système unique de produits stœchiométriquement liés. Dans ce cas, en effet, les vitesses relatives aux réactifs ou aux produits seront proportionnelles entre elles et il suffira d’en étudier plus spécialement une, celle qui est relative au constituant le plus facile à doser, pour définir la vitesse de la réaction:
Le cas d’un système isochore sera également plus facile à étudier que celui d’un système évoluant à volume variable.La réaction isoléeL’étude sera surtout beaucoup plus facile si l’on est en présence d’une réaction chimique isolée, c’est-à-dire conduisant à un système unique de produits stœchiométriquement liés. Dans ce cas, en effet, les vitesses relatives aux réactifs ou aux produits seront proportionnelles entre elles et il suffira d’en étudier plus spécialement une, celle qui est relative au constituant le plus facile à doser, pour définir la vitesse de la réaction: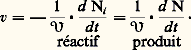 L’objet de la cinétique formelle est de préciser l’influence quantitative de tous les facteurs mesurables sur la vitesse de réaction. Ces facteurs sont en premier lieu les concentrations des constituants, réactifs, produits, catalyseurs... Utilisant les mesures simultanées de la vitesse et des concentrations, on essaiera de déterminer la loi mathématique, dans ce cas une équation différentielle, traduisant au mieux les observations. Quand la réaction isolée est une réaction élémentaire, c’est-à-dire une réaction qui ne comporte qu’un réarrangement chimique, cette loi sera très simple, par exemple:
L’objet de la cinétique formelle est de préciser l’influence quantitative de tous les facteurs mesurables sur la vitesse de réaction. Ces facteurs sont en premier lieu les concentrations des constituants, réactifs, produits, catalyseurs... Utilisant les mesures simultanées de la vitesse et des concentrations, on essaiera de déterminer la loi mathématique, dans ce cas une équation différentielle, traduisant au mieux les observations. Quand la réaction isolée est une réaction élémentaire, c’est-à-dire une réaction qui ne comporte qu’un réarrangement chimique, cette loi sera très simple, par exemple: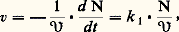 pour une réaction de réarrangement de la molécule se produisant selon un acte monomoléculaire, c’est-à-dire dans lequel aucune des autres molécules présentes n’est impliquée (loi d’ordre un). k est la constante de vitesse. De même, pour une réaction élémentaire impliquant deux espèces chimiques, la loi sera (loi d’ordre deux):
pour une réaction de réarrangement de la molécule se produisant selon un acte monomoléculaire, c’est-à-dire dans lequel aucune des autres molécules présentes n’est impliquée (loi d’ordre un). k est la constante de vitesse. De même, pour une réaction élémentaire impliquant deux espèces chimiques, la loi sera (loi d’ordre deux):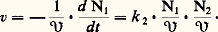 De tels cas sont en fait rares. Par analogie, cependant, on a souvent tendance à adopter a priori une expression de la vitesse qui soit une fonction puissance par rapport à chacun des constituants:
De tels cas sont en fait rares. Par analogie, cependant, on a souvent tendance à adopter a priori une expression de la vitesse qui soit une fonction puissance par rapport à chacun des constituants: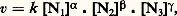 où 見, 廓, 塚 peuvent être entiers ou fractionnaires, positifs ou négatifs, et sont appelés «ordres partiels de la réaction». En fait, cela n’est qu’un formalisme, valable seulement dans un domaine étroit de concentration. Les lois cinétiques d’une réaction isolée, telles qu’elles peuvent être déterminées en observant la vitesse dans une large gamme de concentration, sont plus complexes; ainsi pour la réaction: Br2 + H2 燎 2HBr, réalisée en phase gazeuse, l’expression cinétique trouvée est:
où 見, 廓, 塚 peuvent être entiers ou fractionnaires, positifs ou négatifs, et sont appelés «ordres partiels de la réaction». En fait, cela n’est qu’un formalisme, valable seulement dans un domaine étroit de concentration. Les lois cinétiques d’une réaction isolée, telles qu’elles peuvent être déterminées en observant la vitesse dans une large gamme de concentration, sont plus complexes; ainsi pour la réaction: Br2 + H2 燎 2HBr, réalisée en phase gazeuse, l’expression cinétique trouvée est: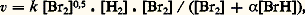 où k et 見 sont deux constantes cinétiques et les quantités [Br2], [H2], [BrH] des concentrations. Ces lois complexes sont dues à ce qu’une réaction isolée est très rarement une réaction élémentaire, mais bien plus souvent résulte d’un enchaînement complexe d’actes élémentaires (cf. chap. 5). La loi cinétique est un des arguments nécessaires à l’élucidation de cet enchaînement, d’où l’importance de la déterminer sous sa forme exacte et explicite.La vitesse de la réaction sera également étudiée en fonction de la température du système. Les constantes de vitesse se modifient avec la température selon une loi, reconnue par Arrhenius, k = k 0e 漣E/RT, où E représente l’énergie d’activation de la réaction. Dans les réactions élémentaires ou qui répondent à un ordre réel vis-à-vis des réactifs, la vitesse suivra une loi semblable. Dans les réactions qui répondent à des lois plus complexes, chaque constante cinétique, k , 見, ..., évolue selon sa propre loi d’Arrhenius, et la variation apparente de la vitesse en fonction de la température n’est plus une simple exponentielle.Cette analyse quantitative réalisée, on dispose de valeurs des constantes cinétiques dont il est très important d’étudier la variation en faisant varier la nature chimique de certains des constituants du système, nature du catalyseur, nature de certains réactifs, nature du milieu réactionnel. Cette dernière partie de l’analyse, souvent qualitative, peut être conduite jusqu’à des relations quantitatives, liant la réactivité chimique à la nature chimique des espèces présentes, à condition de situer les composés chimiques dans des échelles empiriques caractéristiques de telle ou telle propriété et de les représenter ainsi par des valeurs numériques (cf. chap. 6).Les transformations composéesL’étude exhaustive d’une réaction isolée constitue déjà une entreprise de grande ampleur, et dans les travaux usuels on n’en étudie généralement que certains aspects. Or, très souvent, la transformation d’un système chimique passe par un ensemble de réactions; celles-ci sont dites parallèles si les réactifs impliqués dans ces réactions sont indépendants, consécutives si les produits de la première réaction servent de réactifs à la suivante et ainsi de suite, antagonistes si une réaction est contrebalancée par la réaction opposée. Ces transformations sont dites composées ; quand le système de réactions constituant la transformation est plus complexe, on parlera de réactions surcomposées.En fait, la plupart des transformations sont composées et surcomposées. Pour en aborder l’étude, on cherche à réduire la transformation globale en ses composantes que l’on étudiera séparément. Ce but peut être atteint en modifiant le rapport des modes d’activation (catalytique, thermique) lorsque les réactions sont de nature différente, en faisant varier le rapport surface-volume lorsque certaines sont homogènes et d’autres hétérogènes, en s’adressant à divers solvants lorsque les formes actives sont de type différent. On obtient ainsi des équations cinétiques relatives à certaines ou à toutes les réactions impliquées dans la transformation. Cette étude fragmentaire devra être complétée par la formulation d’un modèle mathématique complet, regroupant toutes les équations de vitesse des composantes, et que l’on comparera à l’évolution du système global. Lorsque les artifices pour l’isolement des réactions composantes se sont révélés insuffisants, il faudra établir ce modèle à partir de l’observation globale du système avec mesure simultanée des concentrations de tous les constituants accessibles à la mesure.Nous illustrerons ce point par l’exemple de l’interconversion des butènes entre leurs trois formes isomères, butène-1, cis -butène-2 et trans -butène-2, transformation réalisée sur un catalyseur (fig. 3 a). Tous les équilibres de conversion des butènes deux à deux étant possibles, un modèle mathématique des lois cinétiques a été envisagé (fig. 3 b). Les variations des concentrations en chacun des butènes, partant d’un mélange initial quelconque, ont été mesurées expérimentalement et portées sur le graphique ternaire. On peut déterminer les valeurs des six constantes cinétiques impliquées dans le modèle de façon que l’évolution calculée, traduite par les courbes sur la figure 3 c, englobe parfaitement les points expérimentaux traduisant l’évolution. Ce cas est en fait très simple, mais a l’avantage didactique de se traduire par un graphique unique. Les cas généralement rencontrés sont beaucoup plus complexes, mais on parvient de même, par une étude systématique, à des modèles mathématiques simulant bien les phénomènes et permettant de déterminer les constantes cinétiques des actes élémentaires impliqués par la transformation.5. Schéma et mécanisme réactionnelsLa description par des équations stœchiométriques d’une transformation chimique n’en montre que l’état initial et l’état final. La cinétique doit être capable de décrire l’enchaînement des étapes chimiques menant de l’un à l’autre: c’est le schéma réactionnel ; elle cherche même à décrire comment, dans un acte chimique élémentaire, se produit l’enchaînement des liaisons: c’est le mécanisme réactionnel .Schéma réactionnelLe fait que les réactifs interviennent dans l’équation cinétique par des relations plus ou moins complexes, qui souvent ne sont plus représentables par un ordre, indique bien que la plupart des réactions ne se ramènent pas à un acte unique entre les diverses molécules présentes dans l’équation stœchiométrique, mais se réalisent par un enchaînement d’étapes élémentaires: cet ensemble constitue le schéma réactionnel. Lorsque la réaction se consomme en un acte unique, elle est dite élémentaire et son schéma se réduit à l’équation stœchiométrique. En raison de la faible probabilité pour des partenaires de réactions de se rencontrer, au même instant, dans une disposition géométrique adéquate et avec une énergie suffisante pour réagir, les processus élémentaires ne comportent qu’un nombre réduit de molécules libres, une, deux, et très rarement trois. D’une manière générale, une réaction impliquant plus d’espèces, réactifs, catalyseurs ou initiateurs adjuvants, s’opérera nécessairement par un schéma complexe. Il y a lieu cependant de souligner qu’une stœchiométrie simple ne supposera pas un schéma réactionnel simple; de très nombreux cas le montrent.Les actes chimiques élémentaires n’impliquant qu’un nombre réduit de molécules et ne modifiant qu’un nombre faible de liaisons, il s’ensuit que tout regroupement ou réarrangement de quelque importance doit se produire par une suite de processus qui ne réalisent chacun qu’une transformation minime. C’est le principe du moindre changement de structure. Ce principe s’applique aussi bien à un processus de synthèse qu’à un processus de dégradation: un édifice complexe se désagrégera lui aussi par une succession d’actes élémentaires.Les schémas réactionnels sont innombrables, surtout dans les réactions composées et surcomposées; nous en exposerons deux, très caractéristiques, et que l’on rencontre dans une grande majorité des réactions isolées; ils constituent des voies indirectes pour réaliser la réaction entre deux molécules A + BC + D.Dans le premier schéma, la réaction se décide, et les produits se forment, réellement ou potentiellement, dans une étape unique précédée, elle, par plusieurs étapes d’édification. Il trouve une illustration dans le mode d’action de certains catalyseurs. Dans les stades constructifs, généralement réversibles, le catalyseur K groupe successivement les réactifs pour édifier le complexe réactionnel qui, dans l’étape cinétique, se convertit en une association d’où les produits se dégagent chacun à son tour. Dans un tel schéma, c’est la réaction la plus lente qui constitue l’acte limitatif et règle principalement la vitesse apparente du phénomène. Si l’acte de conversion du complexe AKB est l’acte limitatif, la vitesse sera proportionnelle à sa concentration; celle-ci n’est toutefois pas égale à la concentration analytique en catalyseur K, puisque celui-ci est engagé dans divers complexes. La concentration analytique apparaîtra dans la loi cinétique, mais réduite par une fraction, que l’on appelle facteur de partage de K entre ces diverses formes. Sans être le seul possible, ce schéma intervient dans les divers groupes de catalyses: homogène, hétérogène, enzymatique. À titre d’exemple, le schéma réactionnel de la réaction enzymatique du lactate A, deshydrogéné en pyruvate C sous l’action comme coréactif de la diphosphonucléotide B et d’un enzyme E, est présenté sur la figure 4 ainsi que la loi cinétique observée qui a permis de l’établir. La comparaison des mesures expérimentales et de la loi cinétique permet la détermination des valeurs numériques de la constante de vitesse k r de l’étape limitative et des valeurs des constantes d’association de l’enzyme et des réactifs KEA, KEB, KAEB.Dans le second schéma, la commutation des liaisons chimiques s’effectue par étapes successives par l’intermédiaire de valences libres; il peut être illustré par les réactions en chaînes radicalaires. L’action du brome sur l’hydrogène en est un exemple (fig. 5). Dans une étape désignée comme initiation de la chaîne, les radicaux naissent, dans le cas considéré, sous l’action de la chaleur, ou, dans d’autres cas, sous l’action de la lumière ou d’un initiateur chimique. L’atome de brome soustrait un atome d’hydrogène à la molécule d’hydrogène pour donner une molécule d’acide bromhydrique et un radical hydrogène. Celui-ci réagit à son tour soit sur une molécule de brome, soit sur une molécule d’acide, pour redonner un radical brome et une molécule stable. La répétition du cycle de ces réactions, appelées réactions de propagation, crée une chaîne qui pourrait se propager indéfiniment. En réalité, les atomes et les radicaux se recombinent entre espèces semblables ou différentes et réalisent la rupture de chaîne.L’expérimentation systématique a permis d’établir la loi cinétique, décrite à la figure 5 de façon explicite, et celle-ci a permis de déduire le schéma réactionnel. On y voit apparaître un premier terme, qui traduit le partage du brome entre sa forme moléculaire et sa forme atomique, et une expression fractionnaire qui traduit la probabilité qu’a le radical hydrogène d’attaquer la molécule de brome, formant ainsi le produit cherché, par rapport à l’ensemble des réactions possibles pour ce radical; celle où il attaque une molécule de HBr annule en fait la formation de HBr due à l’action de Br sur H2; elle est donc inopérante.Dans des réactions composées et surcomposées, de tels schémas s’imbriqueront les uns dans les autres. Dans le cas de transformations hétérogènes, ils seront compliqués du fait de la non-homogénéité de répartition; par exemple, si l’initiation est provoquée par une réaction localisée sur une paroi, la propagation dans la masse du système comportera la migration des espèces actives, celles-ci se régénérant alors par un processus en chaîne.Mécanisme de la réactionPour chaque étape élémentaire de ces schémas complexes, ou pour une réaction élémentaire, même si elle ne représente qu’un changement de liaison minime, il est important d’analyser comment se réalise la commutation des liaisons, c’est-à-dire d’essayer de connaître la disposition des molécules impliquées à tout moment entre l’état initial et l’état final.Faute de pouvoir l’étudier expérimentalement, les chimistes proposèrent d’abord des modèles théoriques pour ces actes élémentaires: la théorie des collisions rattachait la vitesse de l’acte élémentaire à la fréquence des collisions Z0. Cette fréquence dans un acte bimoléculaire est de 3 . 1011 mol-1 . s-1 et correspond assez bien aux vitesses maximales de réactions très rapides telles que les neutralisations acides bases. Pour rendre compte de la lenteur relative de beaucoup d’autres réactions, la théorie affecte cette vitesse d’un facteur de probabilité P donnant le rapport des collisions efficaces par rapport au nombre total. Dans la théorie du complexe activé , la réaction est figurée comme la formation d’un complexe «critique» (Brönsted...) ou activé (Eyring...). Ce complexe noté C+- se décompose avec une vitesse fondamentale liée aux constantes de Boltzmann k et de Planck h. La formule peut s’écrire:
où k et 見 sont deux constantes cinétiques et les quantités [Br2], [H2], [BrH] des concentrations. Ces lois complexes sont dues à ce qu’une réaction isolée est très rarement une réaction élémentaire, mais bien plus souvent résulte d’un enchaînement complexe d’actes élémentaires (cf. chap. 5). La loi cinétique est un des arguments nécessaires à l’élucidation de cet enchaînement, d’où l’importance de la déterminer sous sa forme exacte et explicite.La vitesse de la réaction sera également étudiée en fonction de la température du système. Les constantes de vitesse se modifient avec la température selon une loi, reconnue par Arrhenius, k = k 0e 漣E/RT, où E représente l’énergie d’activation de la réaction. Dans les réactions élémentaires ou qui répondent à un ordre réel vis-à-vis des réactifs, la vitesse suivra une loi semblable. Dans les réactions qui répondent à des lois plus complexes, chaque constante cinétique, k , 見, ..., évolue selon sa propre loi d’Arrhenius, et la variation apparente de la vitesse en fonction de la température n’est plus une simple exponentielle.Cette analyse quantitative réalisée, on dispose de valeurs des constantes cinétiques dont il est très important d’étudier la variation en faisant varier la nature chimique de certains des constituants du système, nature du catalyseur, nature de certains réactifs, nature du milieu réactionnel. Cette dernière partie de l’analyse, souvent qualitative, peut être conduite jusqu’à des relations quantitatives, liant la réactivité chimique à la nature chimique des espèces présentes, à condition de situer les composés chimiques dans des échelles empiriques caractéristiques de telle ou telle propriété et de les représenter ainsi par des valeurs numériques (cf. chap. 6).Les transformations composéesL’étude exhaustive d’une réaction isolée constitue déjà une entreprise de grande ampleur, et dans les travaux usuels on n’en étudie généralement que certains aspects. Or, très souvent, la transformation d’un système chimique passe par un ensemble de réactions; celles-ci sont dites parallèles si les réactifs impliqués dans ces réactions sont indépendants, consécutives si les produits de la première réaction servent de réactifs à la suivante et ainsi de suite, antagonistes si une réaction est contrebalancée par la réaction opposée. Ces transformations sont dites composées ; quand le système de réactions constituant la transformation est plus complexe, on parlera de réactions surcomposées.En fait, la plupart des transformations sont composées et surcomposées. Pour en aborder l’étude, on cherche à réduire la transformation globale en ses composantes que l’on étudiera séparément. Ce but peut être atteint en modifiant le rapport des modes d’activation (catalytique, thermique) lorsque les réactions sont de nature différente, en faisant varier le rapport surface-volume lorsque certaines sont homogènes et d’autres hétérogènes, en s’adressant à divers solvants lorsque les formes actives sont de type différent. On obtient ainsi des équations cinétiques relatives à certaines ou à toutes les réactions impliquées dans la transformation. Cette étude fragmentaire devra être complétée par la formulation d’un modèle mathématique complet, regroupant toutes les équations de vitesse des composantes, et que l’on comparera à l’évolution du système global. Lorsque les artifices pour l’isolement des réactions composantes se sont révélés insuffisants, il faudra établir ce modèle à partir de l’observation globale du système avec mesure simultanée des concentrations de tous les constituants accessibles à la mesure.Nous illustrerons ce point par l’exemple de l’interconversion des butènes entre leurs trois formes isomères, butène-1, cis -butène-2 et trans -butène-2, transformation réalisée sur un catalyseur (fig. 3 a). Tous les équilibres de conversion des butènes deux à deux étant possibles, un modèle mathématique des lois cinétiques a été envisagé (fig. 3 b). Les variations des concentrations en chacun des butènes, partant d’un mélange initial quelconque, ont été mesurées expérimentalement et portées sur le graphique ternaire. On peut déterminer les valeurs des six constantes cinétiques impliquées dans le modèle de façon que l’évolution calculée, traduite par les courbes sur la figure 3 c, englobe parfaitement les points expérimentaux traduisant l’évolution. Ce cas est en fait très simple, mais a l’avantage didactique de se traduire par un graphique unique. Les cas généralement rencontrés sont beaucoup plus complexes, mais on parvient de même, par une étude systématique, à des modèles mathématiques simulant bien les phénomènes et permettant de déterminer les constantes cinétiques des actes élémentaires impliqués par la transformation.5. Schéma et mécanisme réactionnelsLa description par des équations stœchiométriques d’une transformation chimique n’en montre que l’état initial et l’état final. La cinétique doit être capable de décrire l’enchaînement des étapes chimiques menant de l’un à l’autre: c’est le schéma réactionnel ; elle cherche même à décrire comment, dans un acte chimique élémentaire, se produit l’enchaînement des liaisons: c’est le mécanisme réactionnel .Schéma réactionnelLe fait que les réactifs interviennent dans l’équation cinétique par des relations plus ou moins complexes, qui souvent ne sont plus représentables par un ordre, indique bien que la plupart des réactions ne se ramènent pas à un acte unique entre les diverses molécules présentes dans l’équation stœchiométrique, mais se réalisent par un enchaînement d’étapes élémentaires: cet ensemble constitue le schéma réactionnel. Lorsque la réaction se consomme en un acte unique, elle est dite élémentaire et son schéma se réduit à l’équation stœchiométrique. En raison de la faible probabilité pour des partenaires de réactions de se rencontrer, au même instant, dans une disposition géométrique adéquate et avec une énergie suffisante pour réagir, les processus élémentaires ne comportent qu’un nombre réduit de molécules libres, une, deux, et très rarement trois. D’une manière générale, une réaction impliquant plus d’espèces, réactifs, catalyseurs ou initiateurs adjuvants, s’opérera nécessairement par un schéma complexe. Il y a lieu cependant de souligner qu’une stœchiométrie simple ne supposera pas un schéma réactionnel simple; de très nombreux cas le montrent.Les actes chimiques élémentaires n’impliquant qu’un nombre réduit de molécules et ne modifiant qu’un nombre faible de liaisons, il s’ensuit que tout regroupement ou réarrangement de quelque importance doit se produire par une suite de processus qui ne réalisent chacun qu’une transformation minime. C’est le principe du moindre changement de structure. Ce principe s’applique aussi bien à un processus de synthèse qu’à un processus de dégradation: un édifice complexe se désagrégera lui aussi par une succession d’actes élémentaires.Les schémas réactionnels sont innombrables, surtout dans les réactions composées et surcomposées; nous en exposerons deux, très caractéristiques, et que l’on rencontre dans une grande majorité des réactions isolées; ils constituent des voies indirectes pour réaliser la réaction entre deux molécules A + BC + D.Dans le premier schéma, la réaction se décide, et les produits se forment, réellement ou potentiellement, dans une étape unique précédée, elle, par plusieurs étapes d’édification. Il trouve une illustration dans le mode d’action de certains catalyseurs. Dans les stades constructifs, généralement réversibles, le catalyseur K groupe successivement les réactifs pour édifier le complexe réactionnel qui, dans l’étape cinétique, se convertit en une association d’où les produits se dégagent chacun à son tour. Dans un tel schéma, c’est la réaction la plus lente qui constitue l’acte limitatif et règle principalement la vitesse apparente du phénomène. Si l’acte de conversion du complexe AKB est l’acte limitatif, la vitesse sera proportionnelle à sa concentration; celle-ci n’est toutefois pas égale à la concentration analytique en catalyseur K, puisque celui-ci est engagé dans divers complexes. La concentration analytique apparaîtra dans la loi cinétique, mais réduite par une fraction, que l’on appelle facteur de partage de K entre ces diverses formes. Sans être le seul possible, ce schéma intervient dans les divers groupes de catalyses: homogène, hétérogène, enzymatique. À titre d’exemple, le schéma réactionnel de la réaction enzymatique du lactate A, deshydrogéné en pyruvate C sous l’action comme coréactif de la diphosphonucléotide B et d’un enzyme E, est présenté sur la figure 4 ainsi que la loi cinétique observée qui a permis de l’établir. La comparaison des mesures expérimentales et de la loi cinétique permet la détermination des valeurs numériques de la constante de vitesse k r de l’étape limitative et des valeurs des constantes d’association de l’enzyme et des réactifs KEA, KEB, KAEB.Dans le second schéma, la commutation des liaisons chimiques s’effectue par étapes successives par l’intermédiaire de valences libres; il peut être illustré par les réactions en chaînes radicalaires. L’action du brome sur l’hydrogène en est un exemple (fig. 5). Dans une étape désignée comme initiation de la chaîne, les radicaux naissent, dans le cas considéré, sous l’action de la chaleur, ou, dans d’autres cas, sous l’action de la lumière ou d’un initiateur chimique. L’atome de brome soustrait un atome d’hydrogène à la molécule d’hydrogène pour donner une molécule d’acide bromhydrique et un radical hydrogène. Celui-ci réagit à son tour soit sur une molécule de brome, soit sur une molécule d’acide, pour redonner un radical brome et une molécule stable. La répétition du cycle de ces réactions, appelées réactions de propagation, crée une chaîne qui pourrait se propager indéfiniment. En réalité, les atomes et les radicaux se recombinent entre espèces semblables ou différentes et réalisent la rupture de chaîne.L’expérimentation systématique a permis d’établir la loi cinétique, décrite à la figure 5 de façon explicite, et celle-ci a permis de déduire le schéma réactionnel. On y voit apparaître un premier terme, qui traduit le partage du brome entre sa forme moléculaire et sa forme atomique, et une expression fractionnaire qui traduit la probabilité qu’a le radical hydrogène d’attaquer la molécule de brome, formant ainsi le produit cherché, par rapport à l’ensemble des réactions possibles pour ce radical; celle où il attaque une molécule de HBr annule en fait la formation de HBr due à l’action de Br sur H2; elle est donc inopérante.Dans des réactions composées et surcomposées, de tels schémas s’imbriqueront les uns dans les autres. Dans le cas de transformations hétérogènes, ils seront compliqués du fait de la non-homogénéité de répartition; par exemple, si l’initiation est provoquée par une réaction localisée sur une paroi, la propagation dans la masse du système comportera la migration des espèces actives, celles-ci se régénérant alors par un processus en chaîne.Mécanisme de la réactionPour chaque étape élémentaire de ces schémas complexes, ou pour une réaction élémentaire, même si elle ne représente qu’un changement de liaison minime, il est important d’analyser comment se réalise la commutation des liaisons, c’est-à-dire d’essayer de connaître la disposition des molécules impliquées à tout moment entre l’état initial et l’état final.Faute de pouvoir l’étudier expérimentalement, les chimistes proposèrent d’abord des modèles théoriques pour ces actes élémentaires: la théorie des collisions rattachait la vitesse de l’acte élémentaire à la fréquence des collisions Z0. Cette fréquence dans un acte bimoléculaire est de 3 . 1011 mol-1 . s-1 et correspond assez bien aux vitesses maximales de réactions très rapides telles que les neutralisations acides bases. Pour rendre compte de la lenteur relative de beaucoup d’autres réactions, la théorie affecte cette vitesse d’un facteur de probabilité P donnant le rapport des collisions efficaces par rapport au nombre total. Dans la théorie du complexe activé , la réaction est figurée comme la formation d’un complexe «critique» (Brönsted...) ou activé (Eyring...). Ce complexe noté C+- se décompose avec une vitesse fondamentale liée aux constantes de Boltzmann k et de Planck h. La formule peut s’écrire: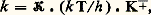 où k T/h a la dimension d’une fréquence et pour valeur numérique 6 . 1012 s size=1漣1 à 300 K, 留 est un coefficient de probabilité et K+- la constante d’équilibre entre le complexe activé C+- et les réactifs initiaux.Quoique l’image et le langage de cette dernière théorie soient largement appliqués et utiles, car ils représentent bien les étapes constructives de la réaction, il faut bien admettre que tous les progrès réels découlent des études expérimentales qui s’efforcent de préciser la structure exacte des états intermédiaires entre état initial et état final. Dans ce cadre, diverses techniques ont permis de grands progrès: la stéréochimie qui, en précisant la position relative dans l’espace des liens des molécules initiales et finales, permet de définir par quelle «face» d’une molécule a lieu l’approche du coréactif. Les effets isotopiques , ou modification de la vitesse d’une réaction obtenue en remplaçant un hydrogène d’une molécule par un deutérium, indiquent si cet hydrogène est directement mobilisé par la réaction. L’étude de réactions très rapides , réactions de protonation par exemple (A + H+AH+), va enfin permettre la détermination expérimentale exacte de la vitesse des actes les plus élémentaires de la chimie; ces études n’ont été possibles que par l’utilisation de matériel d’étude à réponse ultra-rapide pour suivre soit des réactions, soit des retours à l’équilibre de système perturbé par une excitation très brève (phénomènes de relaxation). Ce sont des travaux de cet ordre qui ont été distingués par l’attribution du prix Nobel 1967 à R. G. W. Norrish, G. Porter et M. Eigen.6. Structure chimique et réactivité ; échelles de réactivitéLa multitude des substances chimiques, la multiplicité des systèmes réactionnels qui en dérivent donnent lieu à un nombre incalculable de réactions chimiques. Il importe donc de dégager des résultats expérimentaux des informations pour prévoir le comportement réactionnel des espèces chimiques. Si, dans un système chimique donné, on ne varie, toutes choses égales d’ailleurs, que la nature chimique d’une partie R d’un des constituants RX du système, on dit que l’on réalise une famille de réactions chimiques I par rapport au réactif RX. On mesure alors pour chaque système de la famille la valeur de la vitesse de réaction et on dispose d’une correspondance entre la nature des réactifs modifiés (R0, R1, R2, ..., Rn ) et une série de chiffres de vitesse (k I, 0, ..., k I, n ). Si la réaction est élémentaire, ces valeurs doivent dépendre de façon assez directe de la nature de R. Considérons une deuxième famille de réaction II prise, par rapport à la même suite de réactifs RY, Y étant ou non semblable à X, engagés dans un système chimique II différent du système I par exemple par la nature d’un coréactif ou du catalyseur. On mesurera une seconde série de constantes de vitesse k II, 0, ..., k II, n . Si les deux réactions élémentaires étudiées sont régies par un mécanisme assez semblable, les deux séries de chiffres pourront être reliées par une loi de similitude dont il s’agit de trouver la forme mathématique; il faut d’abord considérer les suites de chiffres en valeur relative par rapport à l’un des termes, celui qui correspond au réactif R0 par exemple. Expérimentalement, Brönsted, puis surtout Hammett ont montré que cette loi était de la forme:
où k T/h a la dimension d’une fréquence et pour valeur numérique 6 . 1012 s size=1漣1 à 300 K, 留 est un coefficient de probabilité et K+- la constante d’équilibre entre le complexe activé C+- et les réactifs initiaux.Quoique l’image et le langage de cette dernière théorie soient largement appliqués et utiles, car ils représentent bien les étapes constructives de la réaction, il faut bien admettre que tous les progrès réels découlent des études expérimentales qui s’efforcent de préciser la structure exacte des états intermédiaires entre état initial et état final. Dans ce cadre, diverses techniques ont permis de grands progrès: la stéréochimie qui, en précisant la position relative dans l’espace des liens des molécules initiales et finales, permet de définir par quelle «face» d’une molécule a lieu l’approche du coréactif. Les effets isotopiques , ou modification de la vitesse d’une réaction obtenue en remplaçant un hydrogène d’une molécule par un deutérium, indiquent si cet hydrogène est directement mobilisé par la réaction. L’étude de réactions très rapides , réactions de protonation par exemple (A + H+AH+), va enfin permettre la détermination expérimentale exacte de la vitesse des actes les plus élémentaires de la chimie; ces études n’ont été possibles que par l’utilisation de matériel d’étude à réponse ultra-rapide pour suivre soit des réactions, soit des retours à l’équilibre de système perturbé par une excitation très brève (phénomènes de relaxation). Ce sont des travaux de cet ordre qui ont été distingués par l’attribution du prix Nobel 1967 à R. G. W. Norrish, G. Porter et M. Eigen.6. Structure chimique et réactivité ; échelles de réactivitéLa multitude des substances chimiques, la multiplicité des systèmes réactionnels qui en dérivent donnent lieu à un nombre incalculable de réactions chimiques. Il importe donc de dégager des résultats expérimentaux des informations pour prévoir le comportement réactionnel des espèces chimiques. Si, dans un système chimique donné, on ne varie, toutes choses égales d’ailleurs, que la nature chimique d’une partie R d’un des constituants RX du système, on dit que l’on réalise une famille de réactions chimiques I par rapport au réactif RX. On mesure alors pour chaque système de la famille la valeur de la vitesse de réaction et on dispose d’une correspondance entre la nature des réactifs modifiés (R0, R1, R2, ..., Rn ) et une série de chiffres de vitesse (k I, 0, ..., k I, n ). Si la réaction est élémentaire, ces valeurs doivent dépendre de façon assez directe de la nature de R. Considérons une deuxième famille de réaction II prise, par rapport à la même suite de réactifs RY, Y étant ou non semblable à X, engagés dans un système chimique II différent du système I par exemple par la nature d’un coréactif ou du catalyseur. On mesurera une seconde série de constantes de vitesse k II, 0, ..., k II, n . Si les deux réactions élémentaires étudiées sont régies par un mécanisme assez semblable, les deux séries de chiffres pourront être reliées par une loi de similitude dont il s’agit de trouver la forme mathématique; il faut d’abord considérer les suites de chiffres en valeur relative par rapport à l’un des termes, celui qui correspond au réactif R0 par exemple. Expérimentalement, Brönsted, puis surtout Hammett ont montré que cette loi était de la forme: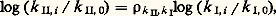 où 福k II,k I est une constante.De cette constatation est venue l’idée de choisir une des suites de chiffres comme échelle d’un caractère de réactivité; ces valeurs de base sont notées 靖 et il existe actuellement plusieurs échelles propres à définir ou à prévoir la réactivité des molécules dans divers types de réactions élémentaires, grâce à des lois dites, selon les cas, de Brönsted, Hammett, Taft, Brown...
où 福k II,k I est une constante.De cette constatation est venue l’idée de choisir une des suites de chiffres comme échelle d’un caractère de réactivité; ces valeurs de base sont notées 靖 et il existe actuellement plusieurs échelles propres à définir ou à prévoir la réactivité des molécules dans divers types de réactions élémentaires, grâce à des lois dites, selon les cas, de Brönsted, Hammett, Taft, Brown...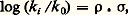 où 靖 est un paramètre caractéristique du réactif et 福 un paramètre caractéristique de la réaction. La figure 6 illustre quelques-unes des premières corrélations observées.En fait, la modification de la structure d’un composé R n’affecte pas seulement sa réactivité, mais également ses autres propriétés, chimiques (aptitude à des associations ou à des dissociations) ou physiques (spectres d’absorption, infrarouge ou ultraviolet, spectre de résonance magnétique nucléaire R.M.N.). Toutes ces mesures expérimentales, quoique diverses, sont liées aux caractéristiques électroniques des composés choisis comme variables. En exprimant à nouveau ces suites de mesures en valeur relative par rapport au composé de base R0 on peut espérer trouver une similitude avec les échelles de réactivité, à condition de choisir une expression mathématique adéquate. C’est ainsi que l’expérience a permis de mettre en évidence des lois:
où 靖 est un paramètre caractéristique du réactif et 福 un paramètre caractéristique de la réaction. La figure 6 illustre quelques-unes des premières corrélations observées.En fait, la modification de la structure d’un composé R n’affecte pas seulement sa réactivité, mais également ses autres propriétés, chimiques (aptitude à des associations ou à des dissociations) ou physiques (spectres d’absorption, infrarouge ou ultraviolet, spectre de résonance magnétique nucléaire R.M.N.). Toutes ces mesures expérimentales, quoique diverses, sont liées aux caractéristiques électroniques des composés choisis comme variables. En exprimant à nouveau ces suites de mesures en valeur relative par rapport au composé de base R0 on peut espérer trouver une similitude avec les échelles de réactivité, à condition de choisir une expression mathématique adéquate. C’est ainsi que l’expérience a permis de mettre en évidence des lois: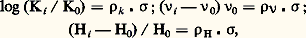 où K est une constante d’équilibre, 益 une fréquence d’absorption, H un déplacement chimique mesuré en R.M.N. Toutes ces valeurs relatives que l’on arrive ainsi à rapprocher des valeurs 靖 traduisent l’effet différentiel que la variation de structure de R provoque sur une différence d’énergie, énergie d’activation nécessaire pour provoquer la réaction chimique, énergie chimique entre deux états en équilibre, énergie nécessaire à un transfert électronique...L’importance de ces études, qui ne progressent que lentement en raison du très grand nombre de données expérimentales nécessaires et de la difficulté à choisir les échelles 靖 les plus caractéristiques pour tel ou tel caractère de la molécule, est donc de permettre de représenter les espèces chimiques par des valeurs scalaires, traduisant leur comportement dans un certain nombre de situations: emploi comme réactif dans un système chimique, comme solvant d’un système réactionnel, ou comme agent d’absorption ou d’émission électromagnétique. Ces valeurs scalaires feront peu à peu partie de la fiche d’identité des espèces chimiques au même titre que des températures de fusion ou des points d’ébullition.7. Applications techniquesMais tout en contribuant aux progrès mêmes de la chimie en tant que science, la cinétique chimique est aussi une science appliquée; en traduisant la transformation chimique en termes quantitatifs, elle rend possible l’action de l’ingénieur sur celle-ci. Un premier groupe d’application concerne la mise au point des procédés industriels de transformation chimique. Le problème dans ce cas est de fabriquer un certain produit chimique au coût minimal. Si nous considérons le cas du Nylon, il faudra disposer d’acide adipique, C2H(CH2)4C2H, au moindre prix, or celui-ci peut être obtenu à partir de benzène, C6H6, hydrogéné en cyclohexane, C6H12, lui-même oxydé par l’air en cyclohexanol, celui-ci étant oxydé par l’air ou par l’acide nitrique en acide adipique. Chacune de ces étapes réactionnelles doit être industriellement optimalisée, c’est-à-dire qu’il faut en faire l’étude cinétique complète, en déduire un schéma de procédé pour une réalisation industrielle, schéma qui définira les fractionnements nécessaires pour purifier les réactifs, les réacteurs nécessaires pour assurer la transformation, les fractionnements nécessaires pour purifier les produits ainsi que les connexions entre ces équipements. On établira ensuite le plan de circulation des fluides, c’est-à-dire qu’en tout point on établira la concentration et le débit pour tous les produits chimiques présents. À partir de ces données, on établira un schéma d’instrumentation et de tuyauteries où sont déterminés les matériaux, les dimensions et les épaisseurs des réacteurs, des colonnes de fractionnement et des tuyauteries... Même une réaction aussi simple que l’hydrogénation du benzène en cyclohexane (C6H6 + 3H2 C6H12) nécessite une installation chimique d’apparence complexe comme le montre la photo ci-contre; la cinétique chimique, la physico-chimie, le génie chimique sont les sciences principales qui permettent, malgré cette complexité, de rendre disponibles les produits chimiques de synthèse à des prix de plus en plus faibles.Une deuxième classe d’application concerne la stabilisation des matériaux; la dégradation d’un matériau est souvent chimique, due en particulier à l’oxydation par l’air, à l’action de l’eau ou à l’action des rayons ultraviolets provoquant des réactions photo-induites dans le matériau. Il est important de déterminer quantitativement quels ingrédients, incorporés au matériau sous une forme et à une concentration précises, rendront ces réactions les plus lentes possibles. Ainsi le polyéthylène peut être employé dans des objets innombrables, parce qu’on a su le stabiliser par des antioxygènes, en général des phénols substitués engagés à raison de 0,1 à 0,3 p. 100, et par des agents antiultraviolets engagés à des concentrations de 0,05 à 0,1 p. 100 et qui souvent sont des dérivés du benzothiazol ou de la benzophénone.Une troisième classe d’application se rattache à l’ensemble des sciences de la vie. Les mécanismes des cellules vivantes apparaissent nettement, depuis que toutes les espèces chimiques présentes ont pu être chimiquement caractérisées, c’est-à-dire surtout depuis 1940, comme obéissant aux lois de la chimie: «Cells obey the laws of chemistry » (J. D. Watson, Molecular Biology of the Gene ). À une mise en évidence qualitative et globale des mécanismes chimiques correspondant à telle ou telle fonction succède de plus en plus une compréhension fine et quantitative. L’attribution du prix Nobel 1967 de médecine à G. Wald pour ses travaux sur les fondements moléculaires de la vue, en particulier sur les mécanismes chimiques au niveau de la rétine, est significative de l’importance attachée à ces travaux. La connaissance quantitative des phénomènes biochimiques s’accompagne en effet d’une possibilité d’action accrue dans les domaines relatifs au contrôle de la reproduction et de la croissance des espèces végétales ou animales, ou au domaine de la médecine. Les agents chimiques, aliments, médicaments, engrais, pesticides, pourront en effet être utilisés de façon de plus en plus précise, et en quantité et en temps, pour leur conférer l’efficacité maximale dans leur interaction avec les phénomènes biochimiques qu’ils ont pour but de contrôler. Là encore, c’est en traduisant la réalité chimique en termes logiques et sous forme de lois quantitatives que la cinétique chimique augmente l’efficacité des techniciens.
où K est une constante d’équilibre, 益 une fréquence d’absorption, H un déplacement chimique mesuré en R.M.N. Toutes ces valeurs relatives que l’on arrive ainsi à rapprocher des valeurs 靖 traduisent l’effet différentiel que la variation de structure de R provoque sur une différence d’énergie, énergie d’activation nécessaire pour provoquer la réaction chimique, énergie chimique entre deux états en équilibre, énergie nécessaire à un transfert électronique...L’importance de ces études, qui ne progressent que lentement en raison du très grand nombre de données expérimentales nécessaires et de la difficulté à choisir les échelles 靖 les plus caractéristiques pour tel ou tel caractère de la molécule, est donc de permettre de représenter les espèces chimiques par des valeurs scalaires, traduisant leur comportement dans un certain nombre de situations: emploi comme réactif dans un système chimique, comme solvant d’un système réactionnel, ou comme agent d’absorption ou d’émission électromagnétique. Ces valeurs scalaires feront peu à peu partie de la fiche d’identité des espèces chimiques au même titre que des températures de fusion ou des points d’ébullition.7. Applications techniquesMais tout en contribuant aux progrès mêmes de la chimie en tant que science, la cinétique chimique est aussi une science appliquée; en traduisant la transformation chimique en termes quantitatifs, elle rend possible l’action de l’ingénieur sur celle-ci. Un premier groupe d’application concerne la mise au point des procédés industriels de transformation chimique. Le problème dans ce cas est de fabriquer un certain produit chimique au coût minimal. Si nous considérons le cas du Nylon, il faudra disposer d’acide adipique, C2H(CH2)4C2H, au moindre prix, or celui-ci peut être obtenu à partir de benzène, C6H6, hydrogéné en cyclohexane, C6H12, lui-même oxydé par l’air en cyclohexanol, celui-ci étant oxydé par l’air ou par l’acide nitrique en acide adipique. Chacune de ces étapes réactionnelles doit être industriellement optimalisée, c’est-à-dire qu’il faut en faire l’étude cinétique complète, en déduire un schéma de procédé pour une réalisation industrielle, schéma qui définira les fractionnements nécessaires pour purifier les réactifs, les réacteurs nécessaires pour assurer la transformation, les fractionnements nécessaires pour purifier les produits ainsi que les connexions entre ces équipements. On établira ensuite le plan de circulation des fluides, c’est-à-dire qu’en tout point on établira la concentration et le débit pour tous les produits chimiques présents. À partir de ces données, on établira un schéma d’instrumentation et de tuyauteries où sont déterminés les matériaux, les dimensions et les épaisseurs des réacteurs, des colonnes de fractionnement et des tuyauteries... Même une réaction aussi simple que l’hydrogénation du benzène en cyclohexane (C6H6 + 3H2 C6H12) nécessite une installation chimique d’apparence complexe comme le montre la photo ci-contre; la cinétique chimique, la physico-chimie, le génie chimique sont les sciences principales qui permettent, malgré cette complexité, de rendre disponibles les produits chimiques de synthèse à des prix de plus en plus faibles.Une deuxième classe d’application concerne la stabilisation des matériaux; la dégradation d’un matériau est souvent chimique, due en particulier à l’oxydation par l’air, à l’action de l’eau ou à l’action des rayons ultraviolets provoquant des réactions photo-induites dans le matériau. Il est important de déterminer quantitativement quels ingrédients, incorporés au matériau sous une forme et à une concentration précises, rendront ces réactions les plus lentes possibles. Ainsi le polyéthylène peut être employé dans des objets innombrables, parce qu’on a su le stabiliser par des antioxygènes, en général des phénols substitués engagés à raison de 0,1 à 0,3 p. 100, et par des agents antiultraviolets engagés à des concentrations de 0,05 à 0,1 p. 100 et qui souvent sont des dérivés du benzothiazol ou de la benzophénone.Une troisième classe d’application se rattache à l’ensemble des sciences de la vie. Les mécanismes des cellules vivantes apparaissent nettement, depuis que toutes les espèces chimiques présentes ont pu être chimiquement caractérisées, c’est-à-dire surtout depuis 1940, comme obéissant aux lois de la chimie: «Cells obey the laws of chemistry » (J. D. Watson, Molecular Biology of the Gene ). À une mise en évidence qualitative et globale des mécanismes chimiques correspondant à telle ou telle fonction succède de plus en plus une compréhension fine et quantitative. L’attribution du prix Nobel 1967 de médecine à G. Wald pour ses travaux sur les fondements moléculaires de la vue, en particulier sur les mécanismes chimiques au niveau de la rétine, est significative de l’importance attachée à ces travaux. La connaissance quantitative des phénomènes biochimiques s’accompagne en effet d’une possibilité d’action accrue dans les domaines relatifs au contrôle de la reproduction et de la croissance des espèces végétales ou animales, ou au domaine de la médecine. Les agents chimiques, aliments, médicaments, engrais, pesticides, pourront en effet être utilisés de façon de plus en plus précise, et en quantité et en temps, pour leur conférer l’efficacité maximale dans leur interaction avec les phénomènes biochimiques qu’ils ont pour but de contrôler. Là encore, c’est en traduisant la réalité chimique en termes logiques et sous forme de lois quantitatives que la cinétique chimique augmente l’efficacité des techniciens.
Encyclopédie Universelle. 2012.